






La enfermedad de Erdheim-Chester es una histiocitosis no-Langerhans. Hasta 2014 han sido reportados alrededor de 550 casos, y está catalogada como enfermedad rara según la European Rare Disease Organization y la National Organization for Rare Disorders.
La clínica más frecuente es dolor óseo en miembros inferiores y generalmente aparece entre la 5.a -7.a década.
El diagnóstico se basa en inmunohistoquímica S100(+/−), CD68(+) y CD1a(−), estos 2 últimos suficientes y mandatorios para el diagnóstico.
El mejor tratamiento consiste en administrar interferón-alfa o interferón-alfa2-pegilado. La supervivencia es del 96% al año y del 68% a los 5 años, siendo menor en casos con afectación del sistema nervioso central.
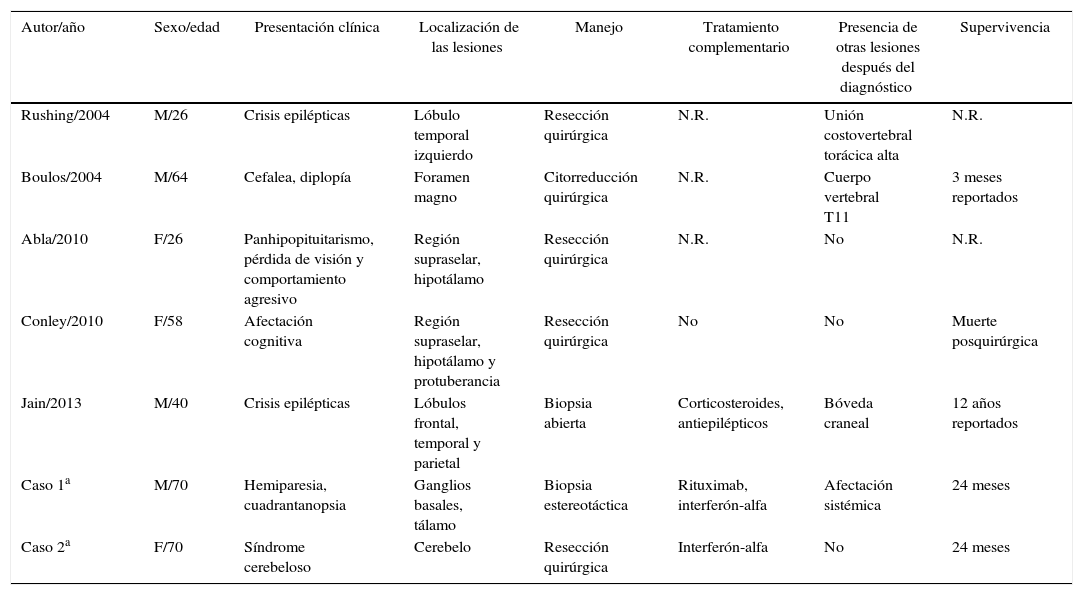
Presentamos 2 casos con afectación aislada del sistema nervioso central al diagnóstico, siendo muy pocos los casos publicados con esta forma de presentación. También observamos que estos pacientes presentaron recidivas o nuevas lesiones a los 8 meses por lo que proponemos seguimiento con RMN cerebral y PET toracoabdominal cada 3-4 meses.
Erdheim-Chester disease is a non-Langerhans histiocytosis. Until 2014 at least 550 cases have been reported. According to European Rare Disease Organization and National Organization for Rare Disorders it is a rare disease.
The most common symptom is bone pain in the lower extremities and it usually appears between the 5th and 7th decades of life.
The diagnostic is based on immunohistochemical results: S100(+/−), CD68(+), and CD1a(−), the latter 2 are mandatory.
The best treatment nowadays is alpha-interferon or pegylated alpha-2.
The overall survival is 96% at one year and 68% at 5 years. Central nervous system involvement is associated with a worse outcome.
Two cases are presentedwith central nervous system lesions in the absence of lesions in other organs on their onset. Very few cases have been reported with this kind of presentation. We also noted that these patients had recurrences or new lesions at 8 months. A follow-up is proposed with brain MRI and thoraco-abdominal PET every 3-4 months.
Artículo
![]()
Si es la primera vez que accede a la web puede obtener sus claves de acceso poniéndose en contacto con Elsevier España en suscripciones@elsevier.com o a través de su teléfono de Atención al Cliente 902 88 87 40 si llama desde territorio español o del +34 932 418 800 (de 9 a 18h., GMT + 1) si lo hace desde el extranjero.
Si ya tiene sus datos de acceso, clique aquí.
Si olvidó su clave de acceso puede recuperarla clicando aquí y seleccionando la opción "He olvidado mi contraseña".




