







Atypical teratoid rhabdoid tumors (AT/RT) of the central nervous system are rare, very aggressive embryological tumors, typically diagnosed in young patients and having a low survival rate after diagnosis. The aim of this study was to emphasize, based on the latest results in the literature, the need for protocols for multidisciplinary treatment in these patients.
Material and methodsWe report our series of 3 cases treated, diagnosed and followed up between 2009 and 2014. They were treated with multimodal therapy protocols (Rhabdoid SIOP-2007 and European Rhabdoid Registry EU-RHAB-2010). In addition, we carried out a literature review.
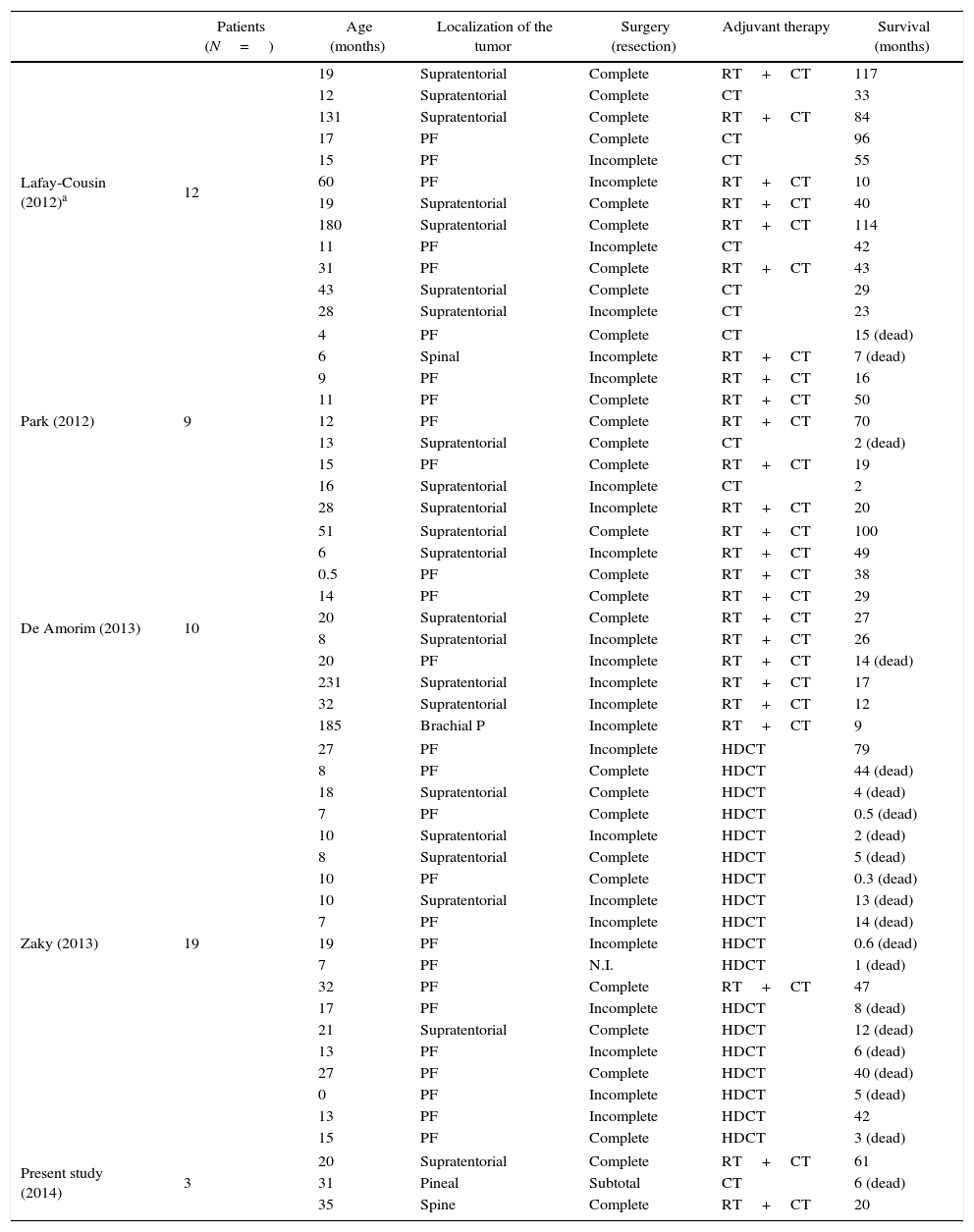
ResultsTwo of our 3 cases (supratentorial and spinal tumors) did not show any progression of the disease after long follow-up, in contrast with most of the cases available in the literature. The second patient had a shorter survival.
ConclusionsPatient age at the time of diagnosis, supratentorial location of the mass and fewer complications with adjuvant treatments seem to be factors yielding good prognosis for AT/RT tumors. In agreement with the latest international protocols, multidisciplinary treatment is the ideal treatment, consisting of radiotherapy and chemotherapy after complete tumor resection.
Los tumores teratoides rabdoides atípicos (TT/RA) del sistema nervioso central son tumores embrionarios muy agresivos, de baja incidencia, típicamente diagnosticados en pacientes jóvenes, con una baja supervivencia tras el diagnóstico. El objetivo de este estudio es resaltar la necesidad del tratamiento multidisciplinar protocolizado de estos pacientes sobre la base de los resultados más actuales de la literatura.
Material y métodosPresentamos nuestra serie de 3 casos de TT/RA diagnosticados, tratados y seguidos entre 2009 y 2014. Nuestros pacientes se trataron siguiendo los protocolos de terapia multimodal (Rhabdoid SIOP-2007, European Rhabdoid Registry EU-RHAB-2010). De forma adicional, se realizó una revisión de la literatura.
ResultadosDos de nuestros pacientes (lesiones supratentorial y espinal) no presentaron progresión de la enfermedad años tras el diagnóstico, en comparación con lo descrito en la literatura. Por otra parte, el segundo paciente presentó una supervivencia menor.
ConclusionesPueden considerarse factores de buen pronóstico: la edad del paciente en el momento del diagnóstico, las lesiones supratentoriales, y las escasas complicaciones de los tratamientos adyuvantes. El consenso actual en cuanto al tratamiento idóneo consiste en cirugía, seguida de quimioterapia y radioterapia.
Article
![]()
If it is the first time you have accessed you can obtain your credentials by contacting Elsevier Spain in suscripciones@elsevier.com or by calling our Customer Service at902 88 87 40 if you are calling from Spain or at +34 932 418 800 (from 9 to 18h., GMT + 1) if you are calling outside of Spain.
If you already have your login data, please click here .
If you have forgotten your password you can you can recover it by clicking here and selecting the option ¿I have forgotten my password¿.



