


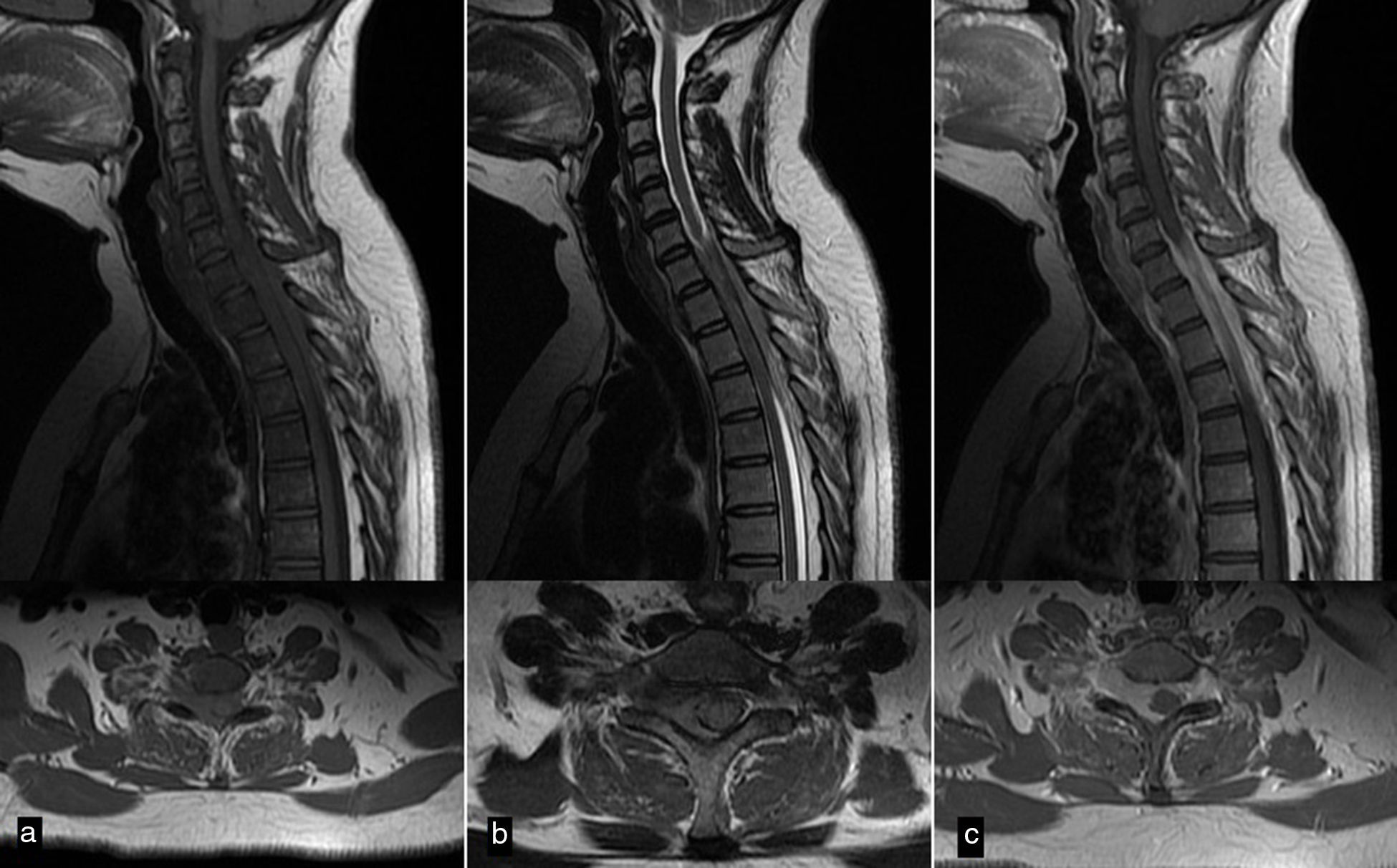
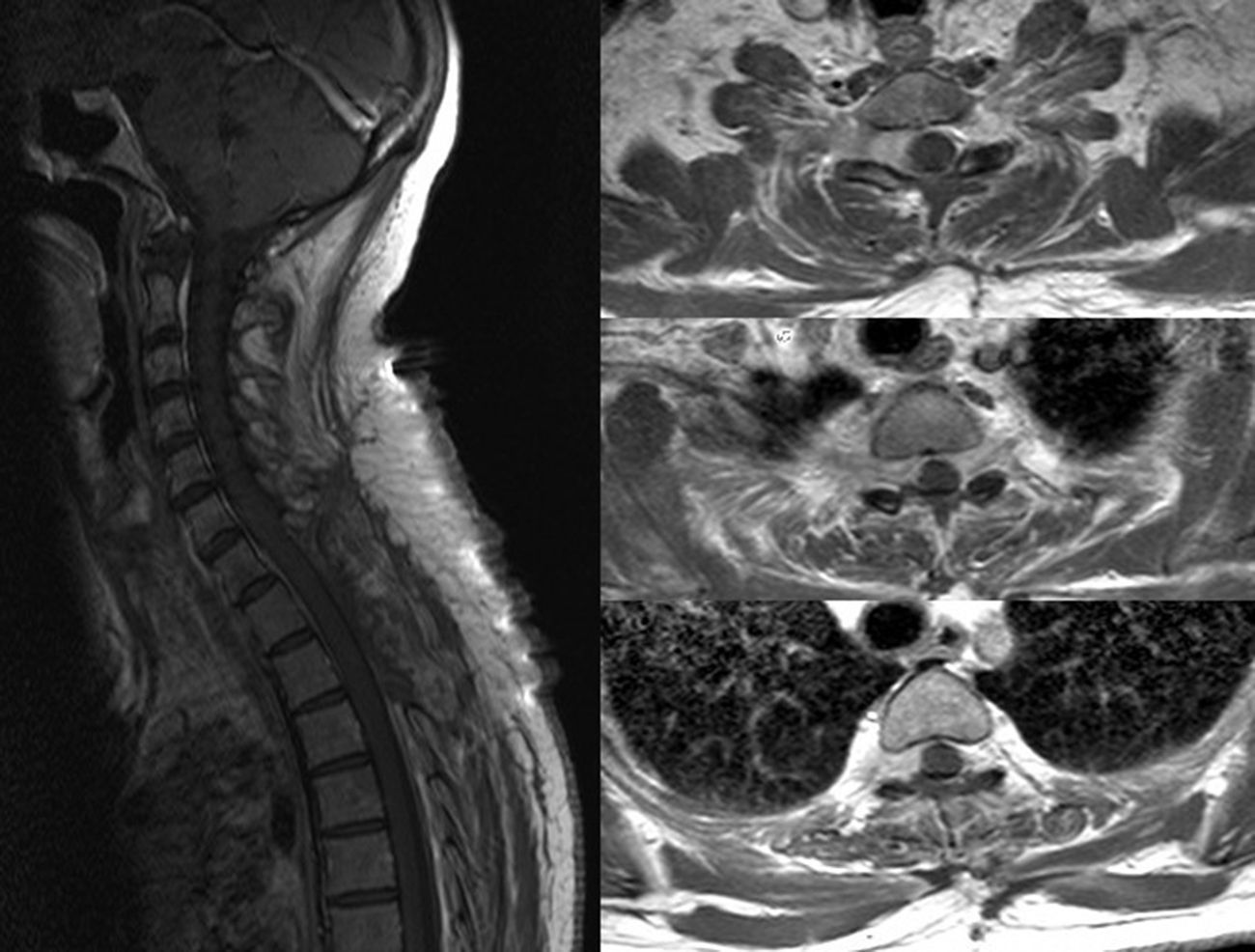
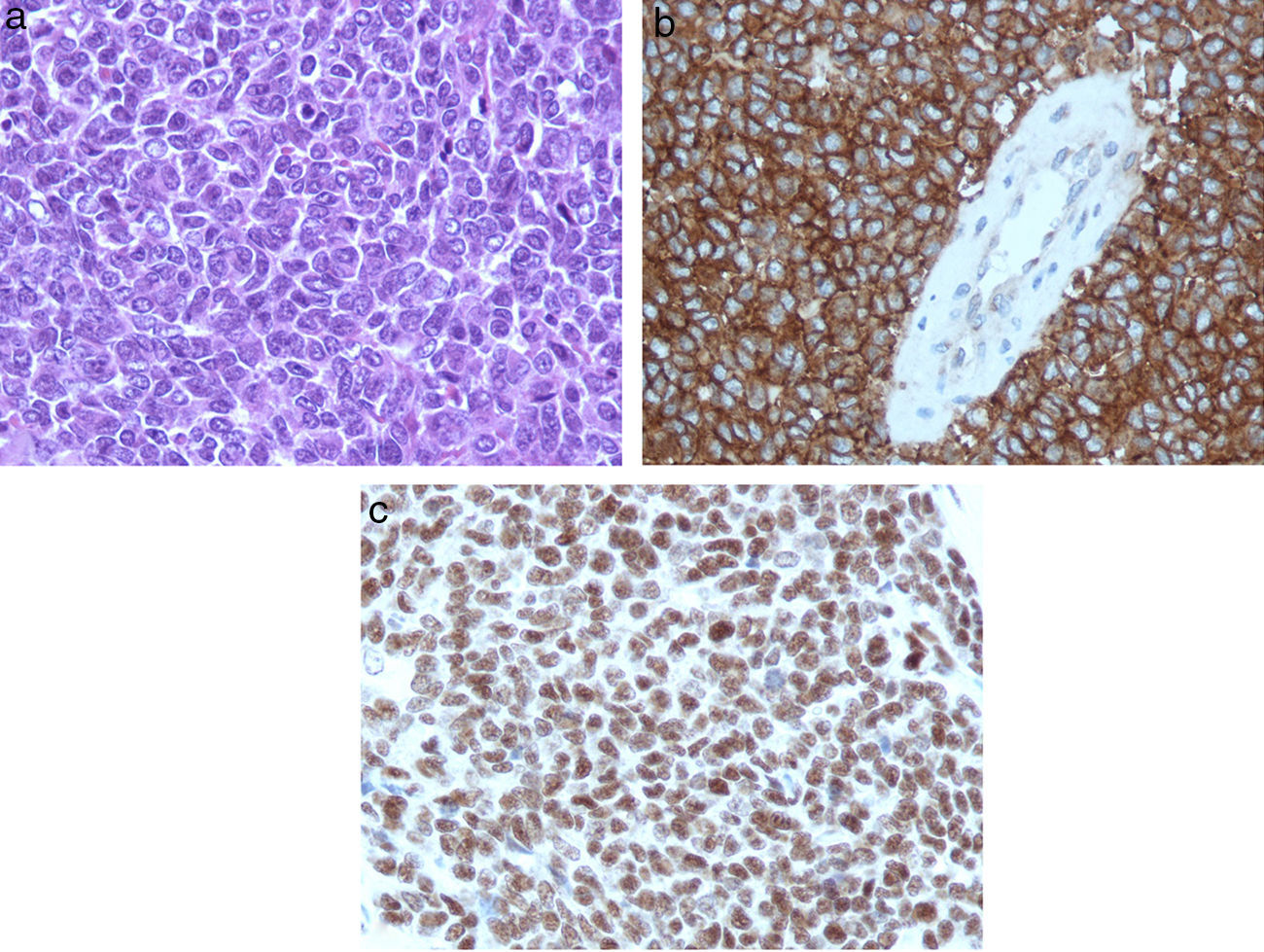
El sarcoma de Ewing es un tumor óseo maligno que en ocasiones presenta afectación extraesquelética, siendo rara la localización epidural. Presentamos el caso de una mujer de 45años que presentó parestesias, debilidad progresiva y retención urinaria. La resonancia magnética mostró una masa epidural desde C6 a D3. Se realizó una laminectomía de C7 a D2 y exéresis parcial de la lesión. El estudio anatomopatológico fue compatible con sarcoma de Ewing. La paciente recibió quimioterapia y radioterapia, no existiendo evidencia de enfermedad a los 8meses de seguimiento. Presentamos una revisión de la literatura de todos los casos publicados de sarcoma de Ewing extraesquelético con afectación epidural.
Ewing sarcoma is a malignant tumour of the bone that sometimes presents extraskeletal involvement, with the epidural location being rare. We report the case of a 45-year-old woman with paresthesia, paresis and urinary retention. Magnetic resonance imaging showed an epidural mass from C6 to D3. Laminectomy from C7 to D2 and partial resection of the lesion was performed. Pathological analysis was consistent with Ewing sarcoma. The patient received chemotherapy and radiotherapy, without evidence of disease at 8 months follow-up. A review of the literature on all published cases of extraskeletal Ewing sarcoma with epidural involvement is presented.
Artículo
![]()
Si es la primera vez que accede a la web puede obtener sus claves de acceso poniéndose en contacto con Elsevier España en suscripciones@elsevier.com o a través de su teléfono de Atención al Cliente 902 88 87 40 si llama desde territorio español o del +34 932 418 800 (de 9 a 18h., GMT + 1) si lo hace desde el extranjero.
Si ya tiene sus datos de acceso, clique aquí.
Si olvidó su clave de acceso puede recuperarla clicando aquí y seleccionando la opción "He olvidado mi contraseña".

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora



