


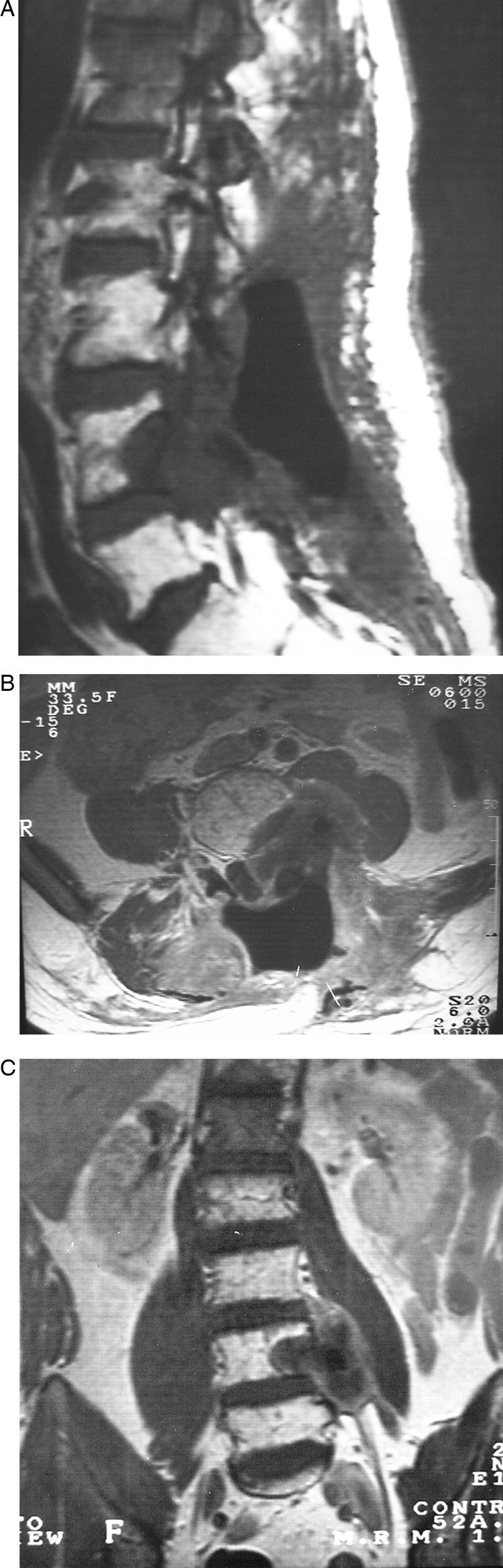
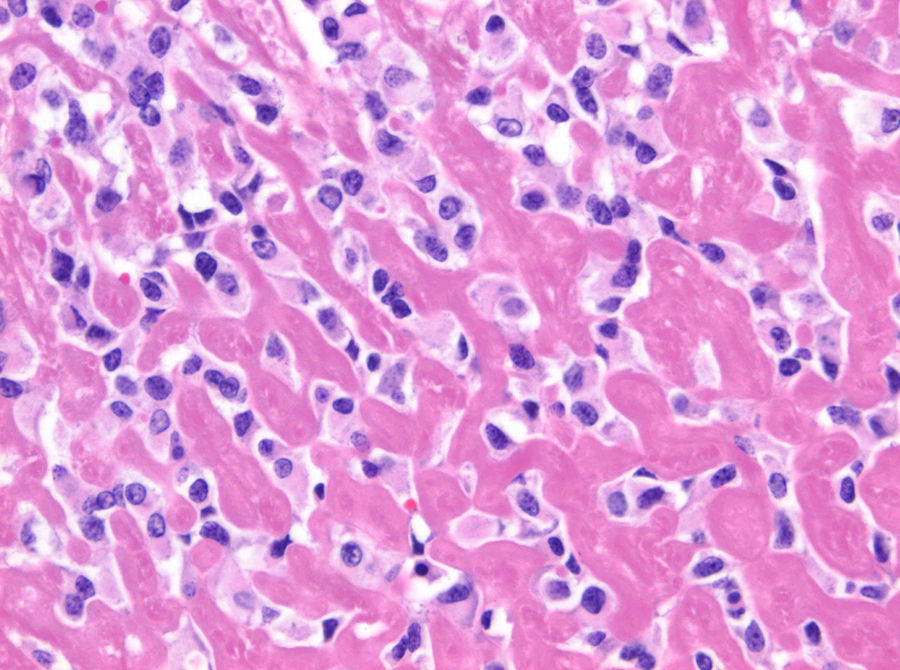
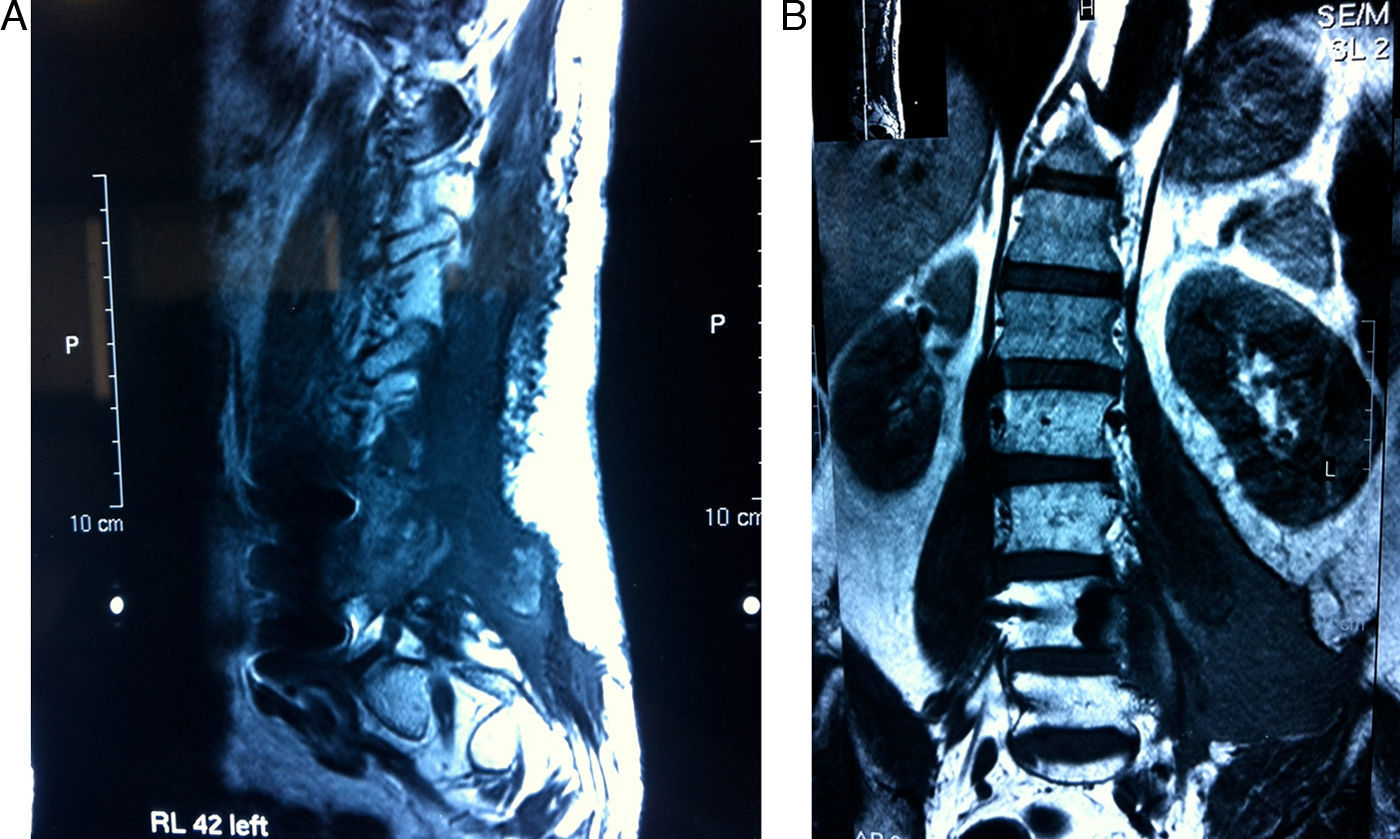
El fibrosarcoma epitelioide esclerosante (SEF) es una variante poco frecuente de fibrosarcoma de bajo grado con características histológicas e inmunohistoquímicas bien definidas, caracterizado por su mal pronóstico. Presentamos un caso de SEF a nivel paraespinal en un paciente varón de 49 años con un tumor que se extiende hacia el foramen L4-L5 e invade la raíz L5. La histología de la pieza quirúrgica y el estudio inmunohistoquímico fueron compatibles con SEF. Este caso es particularmente inusual por su origen a nivel paraespinal e ilustra, a pesar de su bajo grado, el potencial de malignidad del SEF.
Sclerosing epithelioid fibrosarcoma (SEF) is a rare variant of low-grade fibrosarcoma, with specific histological and immunohistochemical features and a poor prognosis. We report a case of SEF of the paravertebral column in a 49-year old male who presented a paraspinal mass with extension into the L4-L5 neural foramen and invasion of the L5 nerve root. Histology of the tumourectomy specimen and its immunohistochemical study led to the diagnosis of SEF. This case was particularly unusual due to its paravertebral column location and, despite its low grade, illustrates the malignant potential of SEF.
Artículo
![]()
Si es la primera vez que accede a la web puede obtener sus claves de acceso poniéndose en contacto con Elsevier España en suscripciones@elsevier.com o a través de su teléfono de Atención al Cliente 902 88 87 40 si llama desde territorio español o del +34 932 418 800 (de 9 a 18h., GMT + 1) si lo hace desde el extranjero.
Si ya tiene sus datos de acceso, clique aquí.
Si olvidó su clave de acceso puede recuperarla clicando aquí y seleccionando la opción "He olvidado mi contraseña".

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora



